Butyl N-acetoxybenzohydroxamate 100a was the first alkyl N-acyloxybenzohydroxamate to be synthesised and was used as the reference compound in the investigation of the mechanism of acid-catalysed solvolysis. Preliminary in-house assays to ascertain the mutagenic potential of this new class of compounds indicated that 100a was significantly mutagenic in the Ames test, with and without metabolic activation. Subsequently, was used as the standard against which the mutagenicity levels and rates of solvolysis for all alkyl N-acyloxybenzohydroxamates were measured.
Butyl N-acetoxybenzohydroxamate 100a was earlier found by Glover et al178 to decompose slowly in aqueous acetonitrile to a mixture of products indicating quantitative formation of acetic acid. The progress of the reaction was monitored using 1H NMR analysis by automatic spectral acquisition at pre-programmed intervals and measuring the disappearance of the acetoxy methyl peak at d2.08. The acetoxy methyl protons were found to be an excellent probe for measuring the rate of reaction due to their isolation from other overlapping resonances. The appearance of acetic acid at d1.95 could also be used as a secondary probe but was not as suitable owing to the overlapping quintet resonance of residual protio-acetonitrile from deuterio-acetonitrile solvent (Figure 2-1).
The progress of solvolsis was determined by measuring the disappearance of the 1H NMR resonance peak for the acetoxy methyl protons at d2.08 through integration and the results are shown in Figure 2-2.
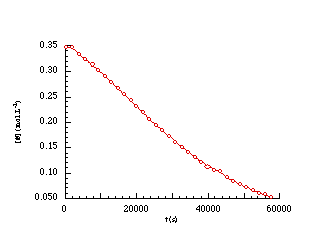
Integration and analysis of the disappearance of 100a in acetonitrile indicated a poor correlation with unimolecular or pseudo-unimolecular kinetics but showed an excellent fit to first-order auto catalysis kinetics for the solvolysis of weak carboxylic esters. The "s" shape to the curve in Figure 2-2 is characteristic of such processes.

Autocatalysis is found where there is an increase in the rate of a reaction through formation of reaction products and is described by the rate equation: rate = k[react][prod]. The small decrease in the pH generated by the release of acetic acid initiates the process and accelerates the rate of solvolysis to a maximum, at which time both acetic and SH+ are at their highest concentration, until the concentration of S (SH+) decreases and the reaction rate slows.
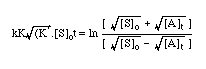 = ln B
EQUATION 2-1
= ln B
EQUATION 2-1The solvolysis of weak carboxylic esters (Scheme 2-4) is described by the integrated equation 2-1 in which [S]o and [A]t are the initial concentrations of butyl N-acetoxybenzohydroxamate 100a and acetic acid at time t respectively, k is the unimolecular or pseudo unimolecular rate constant, K´ is the dissociation constant of acidic acid and K is the pre-equilibrium constant for protonation of butyl N-acetoxybenzohydroxamate. The data from the solvolysis of butyl N-acetoxybenzohydroxamate 100a in aqueous acetonitrile was plotted according to Equation 2-1 and is shown below (Figure 2-3).
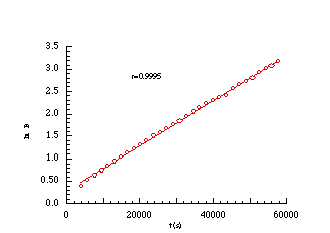
An excellent fit was evident (r2=0.999) and
the composite rate constant (![]() ) at
308K was found to be 8.58x10-5
) at
308K was found to be 8.58x10-5 ![]() but knowledge of the dissociation
constant for acetic acid, K' and the equilibrium constant,
K, under these experimental conditions are required for the
derivation of first-order rate constant, k. These results were
the first to indicate that alkyl N-acetoxybenzohydroxamates
are relatively stable to unimolecular decomposition in
aqueous/organic media.
but knowledge of the dissociation
constant for acetic acid, K' and the equilibrium constant,
K, under these experimental conditions are required for the
derivation of first-order rate constant, k. These results were
the first to indicate that alkyl N-acetoxybenzohydroxamates
are relatively stable to unimolecular decomposition in
aqueous/organic media.
Glover noted that addition of an aliquot of standardised sulphuric acid solution resulted in pseudo first-order acid-catalysed kinetic behaviour.
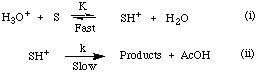
The addition of mineral acid increased the concentration of the protonated substrate, SH+, and increased the observed rate of solvolysis. The half-life of the solvolysis reaction was significantly shortened compared with that for solvolysis without the addition of the mineral acid. Under autocatalytic conditions the rate of solvolysis was determined, in part, by the dissociation of acetic acid, however the addition of sulphuric acid suppressed this pre-equilibrium resulting in pseudo first-order kinetic conditions which simplified the overall rate equation as described above.
The equilibrium equation for protonation of the substrate is given by equation 2-2 which in turn provides the concentration of the protonated intermediate. Substitution gives the rate equation for acid-catalysed solvolysis, 2-3 which can be further simplified to give equation 2-4.
The integrated rate equation for pseudo first-order solvolysis is given in equation 2-6. For acid-catalysed reactions the acid independent rate constant, kH, was calculated by measuring the pseudo-unimolecular rate constant, k', and dividing by the concentration of hydronium ions as indicated in equation 2-5.
In these preliminary experiments and those subsequently performed by the author, the molarity of the standardised sulphuric acid/deuterium oxide solution was determined by standard laboratory procedures.179 The general acid-catalysed solvolysis procedure was to dissolve a small quantity of alkyl N-acetoxybenzohydroxamate 100 in approximately 26% aqueous (D2O) D3-acetonitrile in an NMR tube. An appropriate aliquot of sulphuric acid (D2O) solution was injected to initiate solvolysis and the tube was immediately inserted into the pre-equilibrated probe of the variable temperature NMR spectrometer and allowed to equilibrate at the required temperature before automated spectral analysis was initiated.
A typical acid-catalysed solvolysis reaction for butyl N-acetoxybenzohydroxamate as monitored by 1H NMR is shown in Figure 2-4 and the data, plotted according to equation 2-6, is displayed in Figure 2-5.

The straight line relationship between ln[S]t and time over several half-lives confirmed pseudo first-order behaviour for acid-catalysed solvolysis of butyl N-acetoxybenzohydroxamate. The acid independent rate of solvolysis, kH, was obtained after dividing k' by the concentration of applied hydronium ion in the reaction mixture.
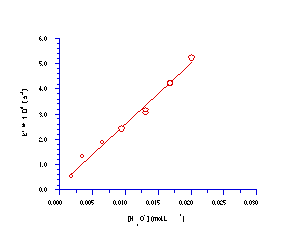
The apparent rate of solvolysis, k´, is linearly dependent upon the concentration of acid and this relationship has been confirmed178 by measuring the change in rate with varying quantities of hydronium ion (Figure 2-6).

Scheme 2.5 also indicates that the acid-independent rate constant for solvolysis, kH, should be inversely proportional to the activity of water. Glover180 et al subsequently showed that the rate of solvolysis is linearly dependent on the inverse of a[H2O] (Figure 2-7).
In summary, initial results in these laboratories indicated that rapid reversible protonation of alkyl benzohydroxamate leads to unimolecular dissociation to acetic acid and products.
A study of the solvent isotope effects178 suggested that the AAl1 rather than the AAc2 process was operating. The rate of solvolysis at different acid concentrations in D2O-CD3CN and H2O-CD3CN varied and the observed solvent KIE was 0.44 ± 0.02, which is consistent with an AAl1 mechanism,180 rather than the AAc2 process for which a larger KIE between 0.48 and 0.69 was predicted.

The unusual AAl1 hydrolysis of t-butyl benzoate esters 102a requires unimolecular ionisation of the protonated intermediated 103a to give a carbocation 105a derived from the alcoholic group (Scheme 2-6). The formation of a charged intermediate is facilitated by polar solvents181 and when the carbocation is tertiary or benzylic.
The Arrhenius parameters for the AAl1 solvolysis of t-butyl ethanoate 102b have been reported.181,182 The entropy of activation for the acid-catalysed process has been determined to be +54 kJmol-1, while the enthalpy of activation was 108 kJmol-1. The positive entropy of activation is consistent with an increase in the disorder of the system coincident with unimolecular dissociation. In contrast, DS for the normal AAc2 process is usually large and negative, indicative of association in the activation step.181
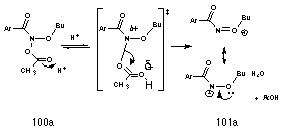
Like tertiary alkyl esters, the mutagens 97 are acetic acid esters with the potential for formation of a stabilised cation, N-acyl-N-alkoxynitrenium ion. By analogy with the solvolysis of t-butyl benzoate esters 102a, the mechanism for nitrenium ion formation from the acid-catalysed solvolysis of butyl N-acetoxybenzohydroxamate 100a was proposed according to Scheme 2-7. Fast, reversible protonation at the carbonyl of the acetoxy moiety increases the leaving potential of the group, resulting in N-OAc bond stretching until insufficient orbital overlap exists and the nitrenium ion 101a and acetic acid separate. The driving force is clearly the increased stability afforded the nitrenium ion by the alkoxy moiety and solvation of this ion by the polar solvent. Such stability has been predicted theoretically by both semi-empirical and ab initio calculations.95,105
Reversible protonation of butyl N-acetoxybenzohydroxamate 100a is in principle possible at the acetoxy and alkoxy ether oxygen, nitrogen, and the carbonyl oxygen of the benzoyl and acetoxy moieties. AM1 calculations, on the model methyl N-acetoxybenzohydroxamate (97 R=CH3, R'=CH3, Ar=C6H5), predict that protonation at the benzamide carbonyl 107 is more favourable than the N-acetoxy carbonyl oxygen 106 by 21 kJmol-1 (Figure 2-8). However, only pre-equilibrium protonation at the acetoxy moiety can lead to ester hydrolysis and nitrenium ion formation.
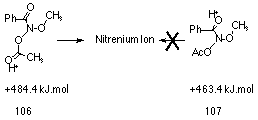
Conclusive evidence for the AAl1 process was obtained in this study from the Arrhenius parameters for solvolysis of butyl N-acetoxybenzohydroxamates as well as substituent effects.
Butyl N-acetoxybenzohydroxamate 100a was solvolysed over the range 298K-338K to determine the acid-independent rate constant, kH, and the resulting Arrhenius data is given in Table 2-1.
|
|
|
|
|
|
|
|
0.05414 |
2.941 |
0.005433 |
-5.2152 |
|
|
0.03248 |
3.650 |
0.01124 |
-4.4886 |
|
|
0.002813 |
0.7229 |
0.02570 |
-3.6614 |
|
|
0.05393 |
75.65 |
0.1403 |
-1.9641 |
|
|
0.002819 |
11.21 |
0.3977 |
-0.9222 |
|
|
0.002781 |
12.26 |
0.4409 |
-0.8190 |
|
|
0.001718 |
28.50 |
1.6596 |
0.5066 |
|
|
0.002808 |
31.928 |
1.1372 |
0.1285 |
|
|
0.001415 |
22.533 |
1.5928 |
0.4655 |
All solvolysis runs displayed excellent pseudo first-order kinetics (r > 0.999) leading to very small errors in k'. The plot of ln kH against the reciprocal of the absolute solvolysis temperature gave an excellent Arrhenius relationship (Figure 2-9).
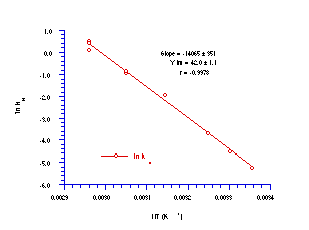
The linear relationship between ln kH and the inverse of absolute temperature indicated that a consistent solvolysis mechanism operated over the temperature range and the Arrhenius parameters for that process were calculated from the slope and intercept. The energy of activation (Ea) was calculated to be 116.9±2.9 kJ mol-1 which is in the region of those cited for the acid-catalysed hydrolysis of tertiary alkyl diphenylmethyl and a-methylallyl esters, namely 120 kJmol-1.181
From the intercept, the Arrhenius plot gives an entropy of activation (DS) of 96.1±9.1 JK-1mol-1 which is even more positive than the ester values (cf. 40 JK-1mol-1 ) and accords with a much looser and probably later transition state with substantial alkoxy-nitrenium ion character. The substantial anomeric overlap between the filled 2p orbital on the alkoxy oxygen and the N-OAc s* orbital accounts for this, since it would facilitate heterolysis of the N-OAc bond. Anomeric effects are strongest in X-N-Y: systems when the Y: lone pair is highest in energy and N-X s* is lowest in energy.121,176,177,181 Relative to RON(COR)OR and CH3COON(COR)OR the protonated intermediate CH3C(OH+)ON(COR)OR would have a much lower O-N s* orbital resulting in a strong anomeric weakening of the bond and a loose transition state.
The Arrhenius data is consistent with alkoxynitrenium ion formation. The transition state would thus involve considerable development of positive charge at nitrogen. The susceptibility of the heterolysis process to electronic factors is reflected in the Arrhenius parameters for solvolysis of a series of butyl N-acetoxy-(para-substituted)benzohydroxamates, 100a-h.
Butyl N-chloro-(para-substituted)benzohydroxamic esters 99b-h, were synthesised via the standard procedure and were converted to the butyl N-acetoxy-(para-substituted)benzohydroxamates 100b-h according to method one, as shown in Scheme 2-3. The compounds were solvolysed in aqueous acetonitrile over the temperature range of 298-338K in the same manner as for the solvolysis of 100a and the Arrhenius parameters are tabulated below (Table 2-2).
|
|
|
|
|
|
|
|
|
|
100b MeO |
43.05±1.29 |
104.7±10.7 |
13592±408 |
113.0±3.4 |
|
|
|
|
100d Me |
44.01±0.13 |
112.7±2.2 |
14429±87 |
120.0±0.7 |
|
|
|
|
100e But |
44.56±1.23 |
117.2±10.2 |
14628±389 |
121.6±3.2 |
|
|
|
|
100c Ph |
44.92±1.06 |
120.6±8.8 |
14834±340 |
123.3±2.8 |
|
|
|
|
100a H |
42.02±1.09 |
96.1±9.1 |
14065±351 |
116.9±2.9 |
|
|
|
|
100g Br |
39.96±0.63 |
79.0±5.2 |
13610±199 |
113.2±1.7 |
|
|
|
|
100f Cl |
39.05±2.37 |
71.4±19.7 |
13331±753 |
110.8±6.3 |
|
|
|
|
100h NO2 |
31.25±0.78 |
6.6±6.5 |
11468±248 |
95.3±2.1 |
|
|
|
Butyl N-acetoxy-(para-substituted)benzohydroxamates 100a-h, all solvolyse with positive entropies of activation, which is consistent with dissociation to acetic acid and nitrenium ions. The slowest compound to solvolyse 100h had the lowest enthalpy of activation (95.3±2.1 kJmol-1) and lowest entropy of activation (6.6±6.5 JK-1mol-1), which combine to give the highest Gibbs Free Energy of activation (93.27 kJmol-1) at 308K. For 100h the entropy change is only marginally positive, indicating that a smaller reorganisation of the ground state is required to generate the transition state geometry. This is confirmed by the low EA, reflecting a relatively small loss of orbital overlap. Data in Table 2-2 shows that electron availability at nitrogen in the transition state is a determining factor. Developing positive charge at nitrogen is consequently stabilised by electron releasing substituents.
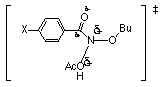
Longer N-OAc bonds for these substituents reduces orbital overlap resulting in higher activation energies. Interestingly, solvolysis must be an entropy driven process as the increase in rate constant, kH, for substrates with electron releasing para substituents is clearly not promoted by the increase in the energy of activation which acts to increase the Gibbs Free Energy of activation. The concomitant rise in the entropy of activation is favourable to the process. The magnitude of �S is consistent with the acid-catalysed solvolysis of t-butyl esters181 and the decomposition of w-diazoacetophenones.183 AAl1 solvolysis of t-butyl mesitoate and t-butyl acetate are 41 and 59 JK-1mol-1 respectively.181
The linear correlation between the activation enthalpy and entropy of activation across a series of related compounds, the so-called isokinetic relationship, has been considered at length by Good et al184 and Exner.185 While the potential artificial nature of the relationship has been debated, it is widely agreed that a linear correlation can be taken as evidence that a similar reaction mechanism is common throughout the series.
With the exception of the p-methoxy substrate 100b, the isokinetic relationship for butyl N-acetoxy-(para-substituted)benzohydroxamates 100a-h displays an excellent correlation between the entropy and enthalpy of activation which indicates that a single mechanism is operating across this series (Figure 2-11). The p-nitro substrate 100h was the slowest compound to solvolyse but had the lowest activation energy and the lowest entropy in the transition state complex with the difference in disorder between the ground state and the transition state configurations only marginally positive. With increasing electron donating substituents, up to p-phenyl 100c, the rise in activation energy of the transition state complex is more than offset by an increase in the entropy of activation.
Butyl N-acetoxy-p-methoxybenzohydroxamate 100b lies somewhat outside the isokinetic line with both the heat of formation and entropy of activation lower than the predicted trend (Figure 2-11).
The tighter transition state and lower activation energy may be due to a non-classical transition state providing a significant, additional supply of electron density to the reactive centre that adds extra stabilisation of the developing positive charge (Figure 2-12). A more orderly transition state with increased alignment between the leaving group, central nitrogen and the ring, would translate into a lower entropy of activation. Tsuno noted a similar discrepancy in the study of the decomposition of w-diazoacetophenones and postulated a similar transition state to account for it.183 In these decomposition reactions, positive charge is also developed a- to a carbonyl.
Clearly in the acid-catalysed solvolysis of 100a-h entropy is the driving force. Electron donating substituents reduce the partial positive charge on the benzamide carbonyl carbon which facilities the formation of the adjacent nitrenium ion. This in turn leads to elongation of the N-OAc bond and an increase in the enthalpy of activation as orbital overlap between N and O decreases. Furthermore the longer N-OAc bond, not withstanding the added constraints imposed by a partially conjugated benzamide moiety, still results in an overall increase in the disorder of the system when electron donating substituents are situated on the ring.
The acid-independent rate constant, kH at 308K was calculated for 100a-h from the Arrhenius parameters and two orders of magnitude difference in rates was evident between the least and most reactive compounds. The rates were correlated with Hammett substituent constants (Figure 2-13).
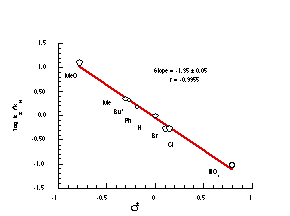
A plot of log(kx/kH) for 100b to 100h displayed a poor correlation with Hammett s constants181,186-191 (r = -0.885) whereas a much better relationship was found using s+ constants (r = -0.995) (Figure 2-13). The negative slope (r= -1.35 0.05) reflects the observed increase in rate when electron donating substituents are present on the ring.
Generally, s+ reactions which involve formation of a positive charge alpha to the ring are significantly influenced by the electronic demands placed on and transmitted through the ring. These reactions tend to have stronger, more negative r values than that observed for solvolysis of 100a-h. For example, the reaction constant for the s+ ionisations of: a)ArCMe2-Cl (90% aq. acetone, 25 oC)190 was -4.45; and b) ArCPh2-OH (H2SO4, 25 oC )192,193 was -3.64. The moderate slope observed from Figure 2-13 arises from the influence of the intervening carbonyl moiety which buffers the reaction centre from the electronic effects of the ring.
For the acid-catalysed solvolysis of 100a-h, a s+ correlation also indicates that the para substituents influence the formation of a developing positive charge at nitrogen through a conjugative interaction. Para substituents can interact directly with the carbonyl carbon thereby increasing or decreasing the positive charge at that centre. Such variations would clearly influence the ease of nitrenium ion formation. The moderate slope of -1.35 is consistent with development of the positive charge beta to the ring, as is the case for nitrenium ion formation. The decomposition of w-diazoacetophenones,183 in which carbenium ion character develops a- to the carbonyl, revealed a comparable Hammett relationship with a negative slope of similar magnitude.
Protonation of the N-acetoxy carbonyl should not be influenced by para substituents on the ring. kH is the composite of k for heterolysis of the N-O bond and K, the pre-equilibrium constant for protonation at the carbonyl. The latter is remote from the influence of para-substituents and K is likely to be similar across the series. Variation in kH is thus largely due to changes in k for the unimolecular step.